

In July of 2014–a little more than two years ago–U.S. Senators Edward Markey, Richard Blumenthal, Elizabeth Warren, Sherrod Brown and Richard Durbin wrote a letter to the FDA telling them to do their job and ensure the validity of laboratory diagnostic tests.
Click to access 2014-07-02_Deese_LDTs.pdf
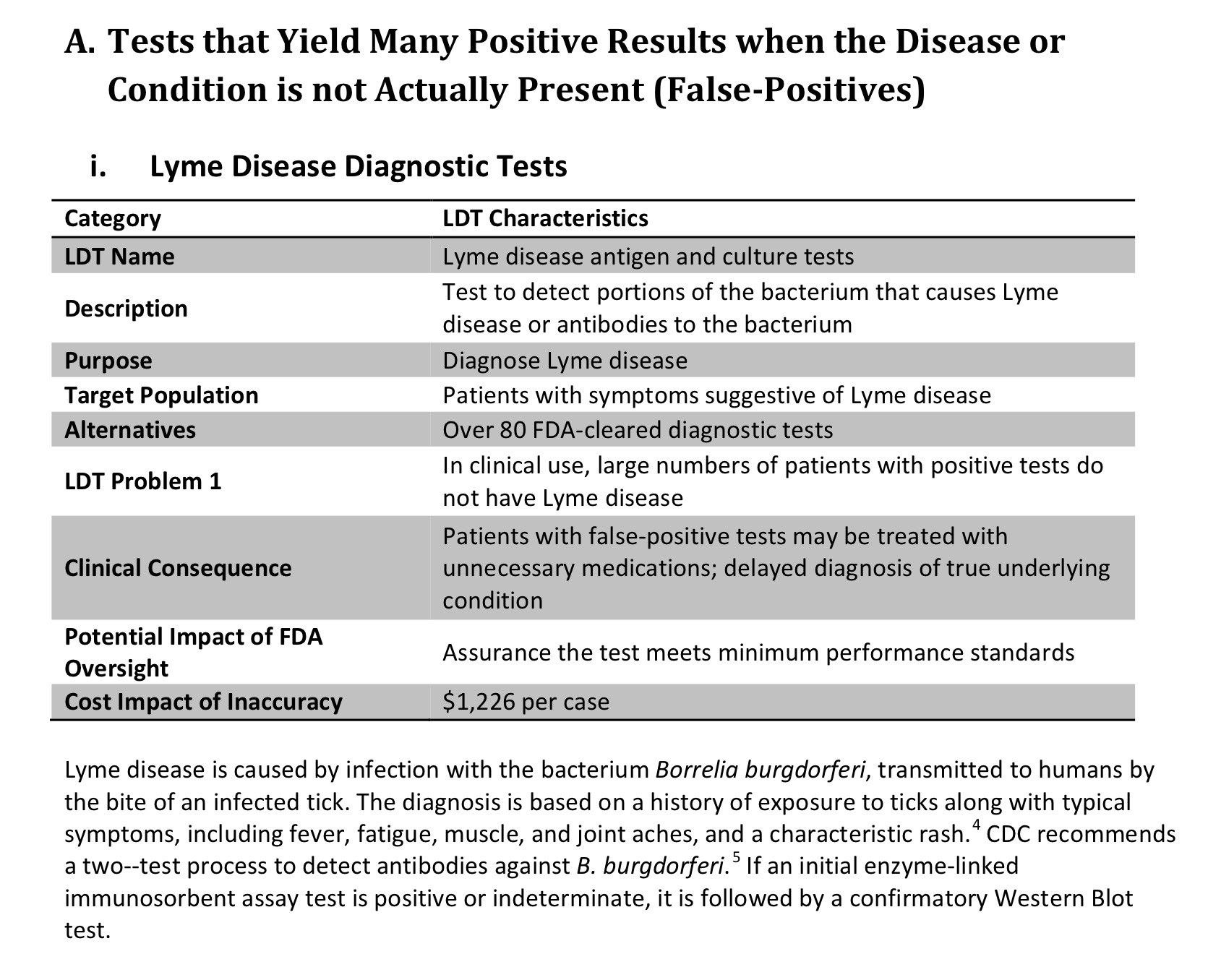
This event inspired our “CDC: No More Tiers” campaign, in which Lyme victims were urged to report their adverse events (such as delay in diagnosis/treatment leading to disability) from the non-scientifically-valid two-tier Lyme testing which is the only testing recognized by the CDC and all of mainstream medicine.
That’s right: the only Lyme test recognized by any government agency, insurance provider or Lexus-driving infectious disease doctor is not scientifically valid–meaning it does not meet the criteria for proving that it detects what it’s supposed to detect.
Here are the basic rules:

So, this guidance was going to be a good thing. Senator Blumenthal, a longtime Lyme advocate, was behind it, and he is well aware of the fraud and racketeering that took place within the CDC, NIH grants office, IDSA, Yale, UConn, etc. regarding the bogus Lyme testing that was put in place at the 1994 CDC Dearborn conference. However, this past Friday, we learned that this FDA guidance has been shelved.
“The US Food and Drug Administration (FDA) on Friday said it will wait for the new administration and halt the finalization of guidance that would have changed the way lab-developed tests (LDTs) are regulated.”
Why the about-face, when the FDA seemed so eager to implement these regulations? They had even notified Congress of their impending actions within 30 days of receiving the senators’ letter. From their notification to Congress:
“Premarket review would ensure that LDTs are properly designed and evaluated for analytical and clinical validity in the intended use population, two critical aspects of IVD performance. Increased oversight through enforcement of the standard device manufacturer adverse event reporting requirements would provide for post-market monitoring of LDTs to assist in identifying any new problems with device performance or quality once the device is in use. Further, appropriate quality controls implemented through compliance with the FDA Quality System regulation (QS reg) (21 CFR Part 820) would lead to more robust and reliable design and manufacture of LDTs with less chance of device defects leading to adverse events.”
And, just so you know I’m not simply being dramatic here, you can see for yourself that Lyme testing was one of the specific examples the FDA provided as to why this regulation was needed. We’ll forgive them for discussing only the “over-diagnosis” of Lyme disease, since the earth truly would spin the wrong direction if they admitted that the real problem is under-diagnosis.
The thing is, it really was all about ensuring validity, or making sure the tests do what they are supposed to do. Sure, it might have called into question some of the tests that are very popular among Lyme patients, but it also would have brought to light the horrible inadequacy of the two-tier Dearborn protocol.
“In contrast, the FDA’s review of analytical validity is done prior to the marketing of the test system, and, therefore, prior to the use of the test system on patient specimens in the clinical diagnosis/treatment context. Further, the FDA’s analytical validity review is more in-depth and more comprehensive than that of the CLIA program, and it is focused on the test system’s safety and effectiveness. As a result, FDA review may uncover errors in test design or other problems with a test system. Also, while CMS’ CLIA program does not address the clinical validity of any test, FDA’s premarket review of a test system includes an assessment of clinical validity.”
While doing my background research for this article I stumbled across a very interesting document. Take a look at what IDSA had to say about this proposed oversight, to the new FDA Commissioner, Robert Califf:
“Dear Commissioner Califf:
The Infectious Diseases Society of America (IDSA), the American Society for Microbiology (ASM), and the Pan American Society for Clinical Virology (PASCV) recognize that the Food and Drug Administration (FDA) is committed to protecting patients. Our societies have closely followed the FDA’s draft guidance proposing to regulate laboratory developed tests (LDTs), and are deeply concerned that the regulations will negatively impact the care of patients being evaluated for infectious diseases (ID)….”
Click to access IDSA%20ASM%20PASCV%20LDT%20letter%20to%20Califf%2004-27-16.pdf
Wait–what?? Infectious disease doctors don’t want accurate testing?
First, as an aside, I think we’re all aware that if pHARMa were a high school, this joker would have been prom king, or at least some superstar jock with a bunch of groupies. His connections run deeper than the average student’s loathing of trigonometry.
“Relationships with the pharmaceutical industry
Califf worked very closely with pharmaceutical companies at the Duke clinical trials center “convincing them to do large, expensive, and, for Duke, profitable clinical trials.”[10] He was a paid consultant for Merck Sharp & Dohme, Johnson & Johnson, GlaxoSmithKline, AstraZeneca, and Eli Lilly per ProPublica from 2009 to 2013. The largest consulting payment was $87,500 by Johnson & Johnson in 2012, and “most of funds for travel or consulting under $5,000”, which has been called “minimal for a physician of his stature”.[11] From 2013-2014 he was paid a total of $52,796, the highest amount was $6,450 from Merck Sharp & Dohme, followed by Amgen, F. Hoffmann-La Roche AG, Janssen Pharmaceutica, Daiichi Sankyo, Sanofi-Aventis, Bristol-Myers Squibb and AstraZeneca.[12] He was the Director of Portola Pharmaceuticals, Inc. from July 2012 to January 26, 2015,[11] Advisor of Proventys, Inc., Chairman of the medical advisory board of Regado Biosciences, Inc. and has been member of the medical advisory board since June 2, 2009, and member of the clinical advisory board of Corgentech Inc.[13] Forbes wrote that his close ties to the drug industry were the reason for him not being nominated for the FDA Commissioner position in 2009.[10]”
https://en.m.wikipedia.org/wiki/Robert_Califf
The IDSA bastards strike again. Not so surprising, really. I mean, it was pretty obvious to anyone paying attention that this could have spelled doom for the current state of Lyme denial. Or was it? Let’s ask Lorraine Johnson, who was last seen:
a. enjoying a margarita at Tres Hombres
b. desperately searching for the IDSA guidelines
c. advocating for patients, and raising their hopes so that when their hopes are dashed again, she will be there to poll them on how it affected their symptoms (i.e. gathering anecdote, or unscientific blah blah that’s good for nothing)
d. all of the above
“When asked what should be done to protect patients if lab tests generated either false negative or false positive results, 85% of patients favored telling patients the risks associated with false test results and permitting patient to make an informed choice regarding whether to take the test. Only 15% believed that tests should be pulled off the market.”
https://www.lymedisease.org/mylymedata/lyme-disease-patients-fda-study/

Right, she was busy collecting anecdote, because, why bother with anything scientifically valid when that would only serve to point out how truly useless you are? Thanks, Lorraine. Glad you and IDSA have something in common. IDSA doesn’t want accurate testing because it would reveal the true magnitude of Lyme disease and the years of deception. Lymedisease-dot-org (or should I say DoD?) doesn’t want accurate testing because the patients are OK with the status quo. Nice. Blame the patients for your screwing them over.
Maybe you can discuss that over mango margaritas on Taco Bowl Tuesday.

Leave a comment